Thanks to a broad range of medical advances, the average American born today is expected to live for about eighty years, in contrast to only fifty years in 1900. For many elderly Americans, however, this welcome increase in life expectancy is marred by a deterioration in cognitive abilities, particularly memory (Fig. 5.5).
Some weakening of memory, beginning around age 40, is normal. Until recently, however, it was not clear whether this age-related memory loss, also called “benign senescent forgetfulness,” is simply the early phase of Alzheimer’s disease or a distinct entity in its own right. The answer to that question is not only a matter of considerable scientific interest, it is also a matter of enormous financial and emotional consequence to our society and its aging population.

Figure 5.5: Prevalence of memory loss in the aging population.
Because implicit and explicit memory are controlled by different systems in the brain, aging affects them differently. Implicit memory is often well preserved in old age, even in the early stages of Alzheimer’s disease. That’s because the disease does not affect the amygdala, the cerebellum, or other areas important for implicit memory until quite late in its course. It also explains why people who are unable to recall the names of loved ones can still ride a bicycle, read a sentence, and play the piano. In contrast, explicit memory—the memory of facts and events—degrades early in people with Alzheimer’s disease.
To find out whether Alzheimer’s and age-related memory loss are biologically different, two groups of scientists at Columbia University collaborated, one led by Scott Small and one by me. We compared three variables: age of onset and progression of each disorder, regions of the brain involved, and r molecular defects in each of the identified regions
To compare age of onset and progression, my colleagues and I turned to mice. Mice do not develop Alzheimer’s disease, but we found that they do show an age-related memory loss that is centered in the hippocampus. This memory loss begins in mid-life, as age-related memory loss appears to do in people. So in mice, at least, we could see that age-related memory loss exists as a separate entity independent of Alzheimer disease.
To find out what areas of the brain are involved in age-related memory loss and what areas are involved in Alzheimer’s disease, Small and his group used brain imaging to study human volunteers ranging in age from 38 to 90. They found, as others had earlier, that Alzheimer’s disease begins in the entorhinal cortex. By contrast they found that age-related memory loss involves the dentate gyrus, a structure within the hippocampus (Small et al., 2015).
Small’s group and mine then collaborated to determine whether the dentate gyrus contains any molecular defects that the entorhinal cortex does not contain. To do this, we examined at autopsy the brain of people between the ages of forty and ninety who did not have Alzheimer’s disease. Using Affymetrix GeneChips, a technology that enabled us to analyze changes in the expression of as many as 23,000 genes, we found 19 gene transcripts that varied with the age of the volunteer. (Transcripts are the single-strand RNA molecules produced in the initial stage of gene expression.) The first and most dramatic change was in a gene called RbAp48. This gene became increasingly less active in the dentate gyrus of older volunteers, resulting in less RNA transcription and less synthesis of the RbAp48 protein. Moreover, the change occurred only in the dentate gyrus, not in any other area of the hippocampus or in the entorhinal cortex.
RbAp48 turned out to be an interesting protein. It is part of the CREB complex, a group of proteins that are critical for the conversion of short-term memory to long term-memory.
Finally, Scott and I returned to mice to see whether expression of the RbAp48 protein also drops off in the dentate gyrus of mice as they age. We found that it does—and once again, the decrease occurs only in the dentate gyrus. In addition, we found that knocking out the RbAp48 gene caused young mice to perform as poorly on spatial tasks as old mice. Conversely, ramping up the expression of the RbAp48 gene in old mice eliminated age-related memory loss, causing them to perform like young mice.
At this point a surprise emerged. Gerard Karsenty, a geneticist at Columbia University, had picked up on the discovery that bone is an endocrine organ and that it releases a hormone called osteocalcin. Karsenty found that Osteocalcin acts on many organs of the body and also gets into the brain, where it promotes spatial memory and learning by influencing the production of serotonin, dopamine, GABA, and other neurotransmitters.
Karsenty and I joined forces to examine whether osteocalcin also affects age-related memory loss. My colleague Stylianos Kosmidis injected osteocalcin into the dentate gyrus of mice and found that it leads to increased PKA, CREB, and RbAp48—the proteins needed for memory formation. Mice that were not given the injections had less CREB and RbAp48. Interestingly, when we gave old mice osteocalcin, their performance on memory tasks such as novel object recognition—which had declined with age—improved. In fact, their memory matched that of young mice. Moreover, osteocalcin even improved the learning capabilities of young mice.
These findings—that osteocalcin declines with age and that it can reverse age-related memory loss in mice—may provide another explanation for the beneficial effects of exercise on the aging human brain. We know that aging is associated with a decrease in bone mass and that the resulting decrease of osteocalcin contributes to age-related memory loss in mice, and possibly in us as well. We also know that vigorous exercise builds bone mass. Thus it is likely that osteocalcin released by the bones ameliorates age-related memory loss in people as well as mice.
Clearly, as these studies illustrate, age-related memory loss is a disorder that is distinct from Alzheimer’s disease—it acts on different molecular processes in a different region of the brain. Moreover, the Roman ideal of a sound mind in a sound body now appears to have a scientific basis.
This is good news for people with a normally aging brain. They can maintain crucial mental functions into old age, provided they eat healthfully, exercise, and interact with others. Just as we have learned to extend the life of the body, we must also extend the life of the mind. Fortunately, as we have seen , several avenues of research encourage us to think that diseases affecting memory including, age related memory loss, may one day be preventable.
It is also important to note that many of the aspects of cognitive function that don’t require memory mature quite well. Wisdom and perspective certainly increase with age. Anxiety tends to decrease. The challenge for all of us is to maximize the benefits of aging while doing our best to minimize the downsides.
Alzheimer’s Disease
Aging seems to target particular areas in the brain, and, as we have seen, the hippocampus is one of the most vulnerable areas. Sometimes it is damaged by lack of blood flow or cell death, but it is often damaged by Alzheimer’s disease.
Alzheimer’s disease is characterized by deficits in recent memory. It results from the loss of synapses, the point of contact where neurons communicate. The brain can regrow synapses in the early stages of the disease, but in the later stages, neurons actually die. Our brain cannot regrow neurons, so this cell death results in permanent damage. Treatment for Alzheimer’s is likely to be most effective early on, before extensive cell death, so neurologists are trying to develop functional brain imaging and other methods of identifying the disease as early as possible.
Scientists have begun to unravel the cascade of events underlying the symptoms of Alzheimer’s disease. They have also learned a great deal about the molecular biology of the disease. Every detail added to that store of knowledge gives us another potential target for a drug, another possible way of halting the progress of this devastating disorder.
The discovery of Alzheimer’s disease dates to 1906, when Alois Alzheimer, a German psychiatrist and colleague of Emil Kraepelin, described the case of a 51-year-old woman, Auguste D., who had become suddenly and irrationally jealous of her husband. Soon thereafter, she developed memory deficits and a progressive loss of cognitive abilities. In time, her memory became so impaired that she could no longer orient herself, even in her own home. She hid objects. She started to believe that people intended to murder her. She was admitted to a psychiatric clinic and died less than five years after the onset of her symptoms.
Alzheimer performed an autopsy on Auguste D and found three specific alterations in the cerebral cortex that have since proven to be characteristic of the disease. First, her brain was shrunken and atrophied. Second, the outside of the nerve cells contained deposits of a dense, plaque-forming material we now know to be an amyloid-beta peptide. Third, inside the neurons was an accumulation of tangled protein fibers that we now call neurofibrillary tangles. Because of the importance of this discovery, Kraepelin named the disorder after Alois Alzheimer.

Figure 5.6: Enhanced photograph of an amyloid plaque and a neurofibrillary tangle in the brain.
Some of what a pathologist sees under a microscope at autopsy we can now see with brain imaging. Figure 5.6 shows the amyloid plaques and neurofibrillary tangles that are hallmarks of Alzheimer’s disease. At first, scientists thought these abnormal protein aggregates were just by-products of the disease, but we now know that they are instrumental in causing it. One of the fascinating things about them is that they form ten to fifteen years before a person’s memory or thinking has begun to change. If these structures could be detected when they first appear—or even before they appear—it might be possible to prevent damage to the brain and to stop Alzheimer’s disease in its tracks.
Plaques initially form in specific, restricted areas of the brain. One such site is the prefrontal cortex As we have seen earlier this part of the brain is involved in attention, self-control, and problem solving. Tangles start in the hippocampus. Amyloid plaques and tangles in these two areas account for the cognitive decline and memory loss in people with Alzheimer’s. At first, the brain is able to compensate well enough that even a family member can’t tell the difference between someone who has this initial damage and someone who does not. Over time, however, as more and more connections are damaged and more neurons begin to die, the brain begins to lose crucial functions. Regions like the hippocampus disintegrate, and symptoms become noticeable.
The Role of Proteins in Alzheimer’s Disease
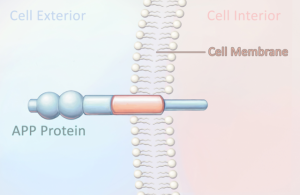
What causes plaques and tangles to form? Scientists have learned that the amyloid-beta peptide is responsible for forming amyloid plaques. This peptide is part of a much larger protein called the amyloid precursor protein (APP), which is thought to be lodged in the cell membrane of dendrites, the short, branching extensions of neurons (Figure 5.7). Two separate enzymes cut through the precursor protein, each in a different place, releasing the amyloid-beta peptide (Fig. 5.8). Once released from the cell membrane, the peptide floats in the space outside the neuron.

Figure 5.7: The amyloid precursor protein (APP) contains the amyloid-beta protein.

Figure 5.8: Two enzymes cut through the amyloid precursor protein lodged in the cell wall: the beta cut, followed by the gamma cut. These cuts release the amyloid-beta peptide (A-beta) into the space outside the cell, where it may form amyloid plaques.
Another protein involved in Alzheimer’s disease is called tau, and it is located inside the neuron. To function, a protein must have a three-dimensional shape. It assumes this shape by means of folding, a process in which the amino acids that make up the protein twist themselves into a very specific conformation. Think of it as exquisitely complicated origami. When a genetic defect causes the tau protein to misfold, it forms toxic clumps (Fig. 5.9) that create neurofibrillary tangles. It turns out that the production and liberation of the amyloid-beta peptide are normal occurrences in everyone’s brain. But in people with Alzheimer’s disease, production of the protein may be accelerated, or clearance of the protein from the area surrounding the cell may be slowed. Either action can result in abnormal accumulations of peptides. What’s more, these peptides are sticky. They adhere to each other and ultimately form the amyloid plaques characteristic of Alzheimer’s disease.

Figure 5.9: A genetic defect causes the tau protein to fold incorrectly. When this happens, the protein clumps inside the cell, forming neurofibrillary tangles.
The combination of these two types of aggregates—plaques outside the nerve cell and tangles within the nerve cell—causes the death of neurons and is responsible for the progression of Alzheimer’s disease.
Genetic Studies of Alzheimer’s Disease
While Alzheimer’s disease usually occurs in people in their 70s or 80s whose families have no history of the disease, a rare, early onset form runs strongly in some families. John Hardy, now at University College London, had the unusual opportunity to study the genetic basis of Alzheimer’s when Carol Jennings got in touch with him.
In the early 1980s Carol’s father was diagnosed with Alzheimer’s disease at age 58. Shortly thereafter, a sister and a brother, both in their mid-fifties, developed the disease. It turns out that Carol’s great-grandfather had had the disease, as had her grandfather and a great uncle. In the main branch of the family, five out of ten children had Alzheimer’s disease, all at the same time. The average age of onset was about 55 (the record for early onset in familial Alzheimer’s is the late 20s).
Hardy and his colleagues wanted to know what genes were inherited by all of the affected siblings in the Jennings family but not by any of the unaffected siblings. They found that the five affected siblings and an affected cousin shared an identical section of chromosome 21, the smallest chromosome in the human genome. But two of the unaffected siblings also had a little bit of that section of chromosome 21. This told Hardy that the gene responsible for Alzheimer’s was not in the bit of chromosome 21 shared with the unaffected siblings. He then looked carefully at the part of chromosome 21 that had been inherited only by the family members with Alzheimer’s, and there he found the defective gene that causes amyloid-beta peptides to clump.
This was the first gene identified in Alzheimer’s disease, and it opened up the study of the disease. Pathologists had already seen that amyloid-beta peptides form plaques, but Hardy showed that in the Jennings family the disease starts with a genetic mutation in the amyloid precursor protein that causes the peptide to clump.
Hardy and other scientists have since found many additional mutations. A group of scientists in Toronto found families with inherited Alzheimer’s who have mutations in the presenilin genes. These mutations prevent presenilin from helping to digest amyloid-beta peptides floating in the space between neurons. This finding fits together beautifully with Hardy’s discovery. Both studies show that all of the families with early onset Alzheimer’s have mutations that lead to amyloid-beta peptides forming deadly clumps in the brain. Put another way, all of the mutations seem to converge on a single common pathway that leads to early onset, familial Alzheimer’s (Fig. 5.10).

Figure 5.10: Several different pathways that lead to early onset Alzheimer’s disease converge on a common pathway to yield a common product: amyloid-beta aggregates. Clusterin is a type of protein that is produced in greater than usual amounts in people with Alzheimer’s disease. It interacts with amyloid-beta to exacerbate the loss of tissue in the entorhinal cortex.
These genetic studies of families with inherited Alzheimer’s led scientists to wonder whether there might be mutations that reduce the number of amyloid-beta peptides. If such mutations exist, do they protect against Alzheimer’s disease?
Thorlakur Jonsson and his colleagues at deCODE Genetics, a biotechnology company in Iceland, have found just such a mutation. It occurs when one amino acid is substituted for another in the amyloid precursor protein, and it results in fewer amyloid-beta peptides being generated. This mutation is particularly interesting because a different amino acid substitution at the same site on that precursor protein causes Alzheimer’s disease. Even more fascinating, people over age 80 who have the protective mutation display better cognitive functioning than people of the same age who lack the mutation.
Risk Factors for Alzheimer’s Disease
Several scientists have been trying to work out the risk factors for the more common, late-onset Alzheimer’s. The most significant risk factor found to date is the Apolipoprotein E (APOE) gene. This gene codes for a protein that combines with fats (lipids) to form a class of molecules called lipoproteins. Lipoproteins package cholesterol and other fats and carry them through the bloodstream. Normal amounts of cholesterol in the blood are essential for good health. Abnormal amounts can clog the arteries and give rise to strokes and heart attacks. One allele, or variation, of this gene is APOE4. The APOE4 allele is rare in the general population, but it puts people at risk of developing late-onset Alzheimer’s disease. In fact, about half of the people with late-onset Alzheimer’s have this allele.
Since we can’t change our genes, is there anything else we can do to lower the risk of developing Alzheimer’s? One possibility has emerged recently, and it has to do with the way our body handles glucose as we age.
Glucose is the body’s main source of energy, and it comes from the food we eat. The pancreas releases insulin, which essentially enables the muscles to absorb glucose. As we age, all of us become a bit insulin-resistant, meaning that our muscles are less sensitive to the effects of insulin. As a result, the pancreas tries to crank out a little bit more insulin, and this makes glucose regulation less stable. If glucose regulation becomes too unstable, we develop type 2 diabetes.
A number of studies have found that type 2 diabetes is a risk factor for Alzheimer’s disease. Furthermore, changes in glucose regulation that accompany type 2 diabetes seem to affect the areas of the hippocampus that are involved in age-related memory loss. The important insight that emerged from these studies is that we can actually modify these age-related changes through diet and physical exercise, which can increase our muscles’ sensitivity to insulin and thus aid in the absorption of glucose.
Environmental factors and comorbidities, or other diseases that people have, may also contribute to the susceptibility to Alzheimer’s disease, but all studies to date point to amyloid clumping as the fundamental cause of dementia. This is a very powerful hypothesis, and it has been extremely useful in guiding research. Recent studies have therefore focused on preventing clumping and clearing pre-existing amyloid clumps by using antibodies that specifically recognize these clumps. As we have seen, disorders such as schizophrenia and depression seem to be caused not by a single gene, but by hundreds of genes, so figuring out how those disorders come about is much more difficult. Even though progress feels slow, our understanding of Alzheimer’s disease has been amazingly rapid.
__
Eric R. Kandel is the University Professor and Fred Kavli Professor at Columbia University, a Senior Investigator at the Howard Hughes Medical Institute, and the recipient of the 2000 Nobel Prize in Phsyiology or Medicine for his studies of learning and memory. His new book, The Disordered Mind: What Unusual Brains Tell Us about Ourselves, is just out from Farrar, Straus & Giroux.
Excerpted from THE DISORDERED MIND: What Unusual Brains Tell Us About Ourselves by Eric R. Kandel, published in August by Farrar, Straus and Giroux. Copyright © 2018 by Eric R. Kandel. All rights reserved.